Targeted Upregulation of PKD1/PKD2 with Programmable PPR Proteins: A Versatile Approach for Addressing Haploinsufficiency
Summary:
Autosomal dominant polycystic kidney disease (ADPKD) is driven largely by insufficient expression of the PKD1 and PKD2 genes, and this study explores a novel RNA-targeted strategy to boost their protein products using engineered pentatricopeptide repeat (PPR) proteins fused to the translational activator eIF4G. By designing PPR-eIF4G constructs that both block inhibitory RNA elements (such as microRNA and uORF sites) and directly recruit translation machinery, the study achieved meaningful increases in PKD1 and PKD2 expression in mammalian cells (up to ~1.6-fold for polycystin-1 and ~3-fold for polycystin-2, levels known from animal models to slow or prevent cyst progression). However, the enhancement was strongly position-dependent: small shifts of only a few nucleotides greatly altered effectiveness, and some constructs increased mRNA without increasing protein, especially at miR-200 sites, highlighting complex post-transcriptional regulation. Overall, these results demonstrate that programmable PPR-eIF4G proteins can precisely and safely upregulate dosage-sensitive genes, offering a promising therapeutic direction for ADPKD and other haploinsufficient disorders.
Download:
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is one of the world’s most common inherited disorders, affecting more than 12 million people. It arises from mutations in the PKD1 and PKD2 genes, which encode the proteins Polycystin-1 (PC1) and Polycystin-2 (PC2). These two proteins are essential for maintaining healthy kidney cell structure and communication. When their levels fall too low, cysts begin to form, eventually leading to progressive kidney failure.
Traditional genetic therapies, while powerful, often come with risks tied to permanent DNA modification. My research explored a more flexible approach, one that enhances the body’s own ability to produce these vital proteins without altering the genome itself.
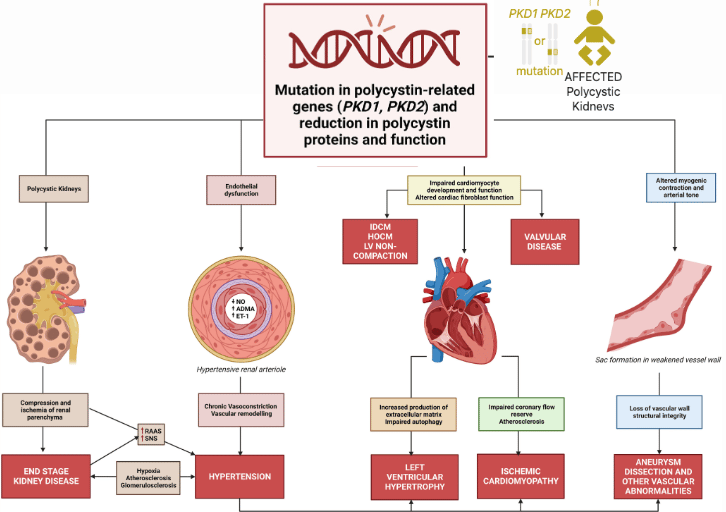
Understanding the Challenge: Autosomal Polycystic Kidney Disease
Pentatricopeptide Repeat Proteins (PPR)
Pentatricopeptide repeat (PPR) proteins are highly programmable RNA-binding proteins that act as precise “molecular readers” of RNA sequences. Built from repeating motifs where each motif recognizes a single nucleotide, PPR proteins can be custom-designed to bind almost any RNA target with exceptional specificity. This modular design allows scientists to attach different functional domains—such as activators of translation, RNA editors, or blockers of inhibitory elements, to control how the cell processes that RNA. Because PPR proteins act on RNA rather than DNA, they offer a reversible, safer alternative to gene-editing tools like CRISPR.
In my research, PPR proteins fused to the translational activator eIF4G are used to selectively bind PKD1 and PKD2 transcripts, enhance their translation, and counteract inhibitory elements, making them a promising platform for therapeutic upregulation in diseases driven by gene haploinsufficiency.
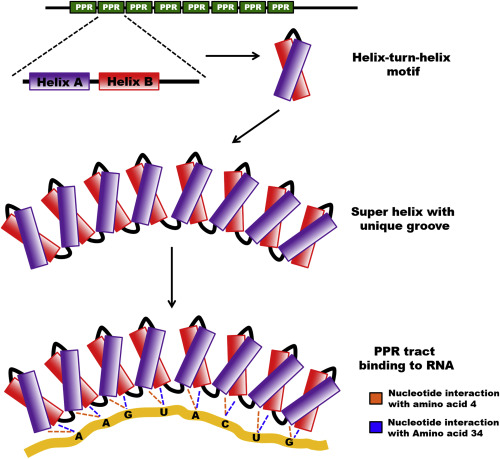
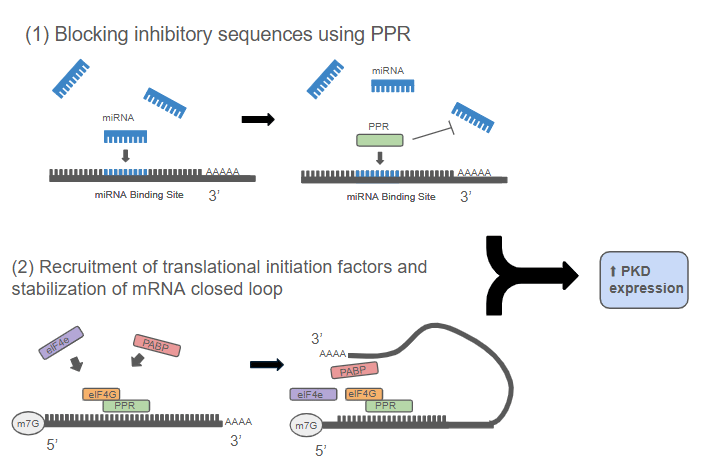
Reprogramming Translation: The PPR-eIF4G System
To boost PKD1 and PKD2 protein production, I engineered synthetic Pentatricopeptide Repeat (PPR) proteins, they are modular RNA-binding proteins capable of recognizing specific mRNA sequences. By fusing these PPRs to eIF4G, a key translation-promoting factor, I designed hybrid proteins that could both block inhibitory sequences and enhance translational efficiency.
This system works through two coordinated actions:
Blocking inhibitory sequences— targeting microRNA binding sites and upstream open reading frames (uORFs) that suppress PKD translation.
Enhancing translation efficiency — recruiting ribosome machinery directly to PKD mRNAs, accelerating the production of Polycystin proteins.
This design not only demonstrated the precision of RNA-level control but also showed how synthetic biology tools can repurpose natural mechanisms for therapeutic goals.
Key Findings: How Much Does PPR Enhance Expression of PKD1/PKD2
Testing these engineered proteins in mammalian cells revealed a significant increase in PKD expression.
PKD1 mRNA rose nearly fourfold, while Polycystin-1 (PC1) levels increased by 1.6×.
Polycystin-2 (PC2) protein abundance grew by about threefold.
These results suggest that even moderate increases in Polycystin levels could meaningfully slow or prevent cyst growth in ADPKD.
Interestingly, the effects depended strongly on where the PPRs bound along the mRNA. Shifting a binding site by just a few nucleotides sometimes led to no translational enhancement, revealing the fine spatial control needed to fine-tune translation.
Another key insight came from studying microRNA-regulated sites, particularly those involving miR-200. In some cases, increased PKD1 mRNA did not lead to a proportional rise in protein, suggesting that microRNAs may influence broader translation mechanisms beyond simple RNA degradation.
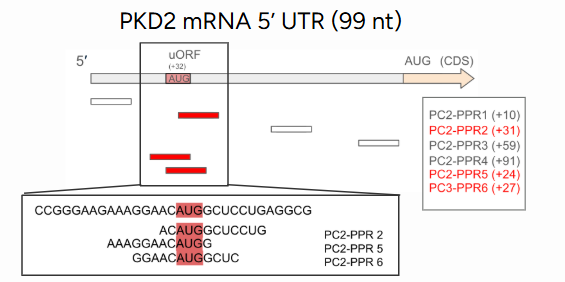
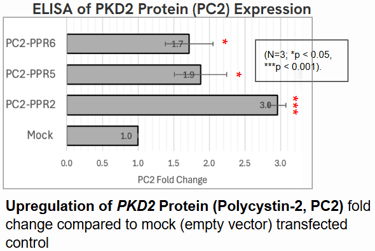
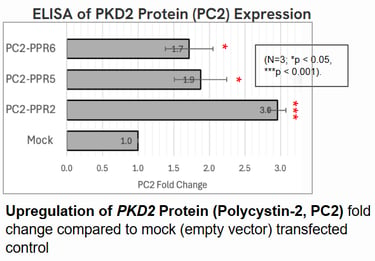
Implications: A Step Toward Gentler Gene Therapy
This work demonstrates that synthetic PPR-based translation enhancement can selectively and reversibly increase protein production in target genes. By acting at the RNA level, this method avoids permanent genome edit, making it potentially safer and more adaptable than traditional gene therapy approaches.
The broader vision is to establish a modular system where customizable PPR proteins could address other genetic diseases caused by haploinsufficiency.
Our team


Genome Chemistry and Engineering Lab
Kyushu University, Japan
Contact
Reach out to discuss ideas or collaborations.
Phone
camtan064@gmail.com
+63 927-114-1655
© 2025. All rights reserved.
Navigation